Thérapeutique
Publié le 07 mar 2019Lecture 10 min
Neuroblastome : caractérisation biologique, traitement actuel et perspectives thérapeutiques
Dominique VALTEAU-COUANET, département cancérologie de l’enfant et de l’adolescent, Hôpital Gustave Roussy, Villejuif
Le neuroblastome est la tumeur solide pédiatrique extra-crânienne la plus fréquente. C’est une tumeur embryonnaire du système nerveux sympathique (médullosurrénale et ganglions sympathiques). Elle survient chez le jeune enfant et présente des caractéristiques cliniques et biologiques très diverses allant de la régression spontanée de très haut risque au pronostic très sévère, malgré des traitements combinés, intenses et prolongés. Cette complexité fait de ces tumeurs un champ de recherche important pour identifier des facteurs pronostics guidant au mieux la thérapeutique et les cibles thérapeutiques qui permettront de développer des traitements plus efficaces dans les formes graves.
Épidémiologie
L’incidence du neuroblastome est de 10,5 par million d’enfants de moins de 15 ans, soit environ 130 à 150 nouveaux cas par an en France. L’âge médian au diagnostic est de 18 mois, 50 % des enfants ayant moins de 1 an au diagnostic et 90 % moins de 5 ans. Les neuroblastomes des adolescents et jeunes adultes sont exceptionnels et de pronostic particulièrement sévère, même si l’évolution de la maladie est souvent prolongée. De rares cas familiaux ont été décrits.
Présentation clinique
La découverte de la maladie est liée soit à des symptômes en rapport avec la tumeur primitive, soit en rapport avec les métastases. La tumeur primitive est développée à partir du système sympathique, elle est abdominale dans 80 % des cas, à point de départ surrénalien pour 50 % des cas, sympathique péri-vasculaire pour 30 % et thoracique dans 15 % des cas. Elle est plus rarement cervicale ou pelvienne. Il arrive exceptionnellement qu’aucune tumeur primitive ne soit retrouvée. La tumeur est découverte soit par les parents ou lors d’un examen systématique par le pédiatre, soit de façon fortuite lors d’un examen fait pour une autre raison – échographie au cours de la grossesse ou à l’occasion de douleurs abdominales. Elle peut aussi se manifester par des symptômes liés à la compression des organes de voisinage, syndrome de Claude Bernard Horner (myosis, ptosis, enophtalmie) dans les tumeurs cervicales, compression médullaire dans les tumeurs présentant un prolongement intra-canalaire (tumeurs dites « en sablier ») et troubles respiratoires pour les tumeurs thoraciques.
Les métastases sont présentes dans environ 50 % des cas et sont alors souvent le point d’appel diagnostic. Elles diffèrent dans leur site en fonction de l’âge des patients. Métastases hépatiques et nodules sous-cutanés chez le nourrisson, elles sont médullaires ou ostéo-médullaires chez l’enfant plus âgé et se manifestent souvent par des douleurs osseuses pouvant entraîner une boiterie. Les métastases rétro-orbitaires sont à l’origine d’hématomes péri-orbitaires et d’une désaxation des globes oculaires (syndrome de Hutchinson). On recherchera des adénopathies à proximité de la tumeur (creux sus-claviculaire). Les métastases pulmonaires et cérébrales sont exceptionnelles au diagnostic. Un syndrome paranéoplasique peut révéler la maladie comme une diarrhée aqueuse en rapport avec une hypersécrétion de peptide vaso-ontestinal (VIP) ou le syndrome opsomyoclonique associant opsoclonies (mouvements rapides multidirectionnels des yeux), ataxie possiblement en lien avec un neuroblastome le plus souvent localisée et de pronostic favorable.
Marqueurs biologiques
Le neuroblastome induit dans plus de 90 % des cas une augmentation des marqueurs urinaires, VMA (acide vanyl-mandélique), HVA (acide homovanillique) et dopamine. Ce dosage doit être réalisé sur les urines recueillies pendant 24 heures. Aucun régime n’est maintenant nécessaire et seules les valeurs rapportées à la créatinine urinaire doivent être prises en compte. Un taux élevé des LDH ou de la NSE peut être identifié. Longtemps considérés comme des marqueurs de mauvais pronostic, ils ne sont pas spécifiques et ne sont pas pris en compte dans la stratégie thérapeutique.
Bilan tumoral
La radiographie du thorax ou l’échographie abdominale constituent souvent les premiers examens réalisés en fonction de la localisation de la tumeur. Ils doivent être complétés par un scanner ou une IRM afin de préciser la localisation de la tumeur, sa taille et la présence d’adénopathies locorégionales. La tumeur est souvent hétérogène avec des calcifications en son sein. Ces examens vont préciser en outre les rapports avec les organes de voisinage et les structures vasculaires. L’objectif est d’apprécier l’existence de facteurs de risque chirurgicaux (IDRFs) et de déterminer si la tumeur est opérable sans risque. Elle sera alors classée L1 en l’absence de métastases associées ou s’il existe des risques vasculaires ou de sacrifice d’organe. Ce qui conduit à la classer L2, selon la classification internationale INRGSS (International Neuroblastoma Risk Group Staging System).
La scintigraphie à la MIBG (méta benzyl guanidine) marquée avec de l’iode 123I MIBG, permet, dans 95 % des cas pour lesquels la maladie capte spécifiquement la MIBG, de faire une cartographie de la maladie et d’identifier s’il existe ou non des métastases (maladie M ou Ms dans la classification INRGSS). Un score a été développé dans le groupe neuroblastome européen SIOPEN pour évaluer l’étendue de la maladie métastatique entre 0 et 72 au diagnostic et son évolution sous traitement. Ce score a une valeur pronostique à la fois au diagnostic et en fin de traitement d’induction avec une limite à 3. La scintigraphie MIBG est un outil indispensable au bilan et à la prise en charge thérapeutique.
L’évaluation de la maladie métastatique est complétée par un bilan médullaire avec des myélogrammes et des biopsies ostéo-médullaires pour un examen cytologique histologique et immuno-histochimique recherchant de cellules neuroblastiques. Au terme de ce bilan, la maladie est classée en stade selon la classification internationale INRG (tableau ci-dessous). Le stade Ms est une forme spécifique au nourrisson de moins de 18 mois, avec la présence de métastases sur les sites cutanés, hépatiques qui dominent généralement le tableau, ainsi que médullaire avec, le plus souvent, un envahissement médullaire minime mais sans envahissement osseux à la MIBG. Ces formes sont de bon pronostic. Enfin, une analyse de la tumeur primitive est réalisée, à partir de la pièce opératoire si une chirurgie est d’emblée décidée après bilan ou biopsie – si possible par voie percutanée. Elle permettra de porter le diagnostic et de faire une classification plus précise si le prélèvement est de taille suffisante, en appréciant le degré de différenciation de la tumeur. L’étude en biologie moléculaire de la biopsie apporte des informations pronostiques essentielles à la prise en charge thérapeutique. L’oncogène NMYC est localisé sur le bras court du chromosome en position 2p24.3. Son amplification identifiée, le plus souvent par des techniques de FISH, est retrouvée dans 20 à 30 % des tumeurs. Elle conduit à les considérer comme de haut risque et à proposer aux patients un protocole intensifié même pour les tumeurs localisées ou métastatiques chez les enfants de moins de 1 an. L’analyse en biologie moléculaire par des techniques de CGH-a ou de SNParray permettent en outre d’identifier des anomalies chromosomiques soit numériques, pertes ou gains de chromosomes entiers associées à un pronostic favorable, soit segmentaires, délétions (1p, 3q, q, 6p ou 11q) ou gains de bras chromosomiques (1q, 2p, 17q), qui sont de pronostic plus défavorable. D’autres anomalies génétiques peuvent être trouvées dans une mutation du gène ALK, présente dans 8 à 10 % des tumeurs et qui pourra dans le futur être ciblée par des thérapeutiques spécifiques.
Prise en charge thérapeutique
Elle dépend du stade de la maladie, de l’âge du patient (les formes de l’enfant de moins de 12 ou 18 mois étant de meilleur pronostic que les formes des enfants plus âgés) et de la biologie de la tumeur, l’existence d’une amplification de l’oncogène NMYC et des anomalies chromosomiques (numériques ou segmentaires). Elle va de l’observation simple à une prise en charge multimodale intensive.
Neuroblastomes de bas risque et de risque intermédiaire
Les nourrissons de moins de 3 mois avec une masse surrénalienne isolée peuvent être surveillés sans bilan spécifique. Les patients avec une maladie localisée sans facteur de risque pour la chirurgie (stade INRG L1) sont opérés et ne nécessitent pas de traitement complémentaire, avec une survie globale de 100 %. Pour les patients avec une maladie localisée L2 de moins de 18 mois ou Ms, le traitement dépend de la présence ou non de symptômes cliniques et/ou du profil génomique de la tumeur. En l’absence de symptômes et avec une biologie favorable de la tumeur, une surveillance simple peut être proposée. Dans les autres cas, une chimiothérapie sera faite en adaptant le nombre de cures et dans l’objectif de la limiter. La chirurgie est faite en l’absence de risque et il n’y a pas d’indication à la radiothérapie. Le pronostic de ces patients est bon avec une survie de 90 %. Les patients avec un neuroblastome de risque intermédiaire stade L2 et âgés de moins de 18 mois sont traités avec une chimiothérapie comportant 6 cures asssociant étoposide et carboplatine ou vincristine, cyclophosphamide et doxorubicine. Les patients ont une chirurgie de la tumeur primitive, une radiothérapie et un traitement d’entretien avec 6 cures d’acide rétinoïque. Leur pronostic doit être amélioré puisque leur survie est < 60 % à 5 ans.
Neuroblastomes de haut risque
Les tumeurs à haut risque sont les tumeurs métastatiques (stade M de la classification INRG) chez des enfants de plus de 1 an et les tumeurs avec une amplification de l’oncogène NMYC quel que soit l’âge du patient.
Le traitement comporte 4 phases :
– une chimiothérapie d’induction dont l’objectif est de faire diminuer le volume de la tumeur primitive et, si possible, de faire disparaitre les métastases ;
– un traitement local comportant une chirurgie de la tumeur primitive et une radiothérapie ;
– une consolidation avec une chimiothérapie à hautes doses (CHD) avec support de cellules souches hématopoïétiques autologues (ASCR) ;
– un traitement d’entretien combinant l’administration d’un anticorps monoclonal anti-GD2 et d’un agent différenciant, l’isorétinoïne (acide rétinoïque).
La chimiothérapie d’induction utilisée dans le groupe SIOPEN est le « Rapid COJEC » ; un traitement comportant 8 cures administrées à 10 jours d’intervalle combinant carboplatine, cisplatine, cyclophosphamide, étoposide et vincristine. Ce traitement a été comparé dans une étude randomisée HRNBL1 R3 au protocole américain qui comportait 5 curesadministrées toutes les 3 semaines combinant cyclophosphamyde, adriamycine et vincristine ou étoposide et cisplatinum. Ce protocole permet d’obtenir les mêmes résultats en termes de réponse métastatique et de survie, avec une toxicité supérieure. Le traitement de consolidation consiste en une chimiothérapie à hautes doses associée à une autogreffe de cellules souches périphériques dont l’objectif est de limiter la toxicité médullaire du traitement. Une étude randomisée menée par le groupe SIOPEN a permis de montrer que la combinaison de busulfan et de melphalan (Bu-Mel) permettait d’augmenter le taux de guérison des patients en comparaison au protocole utilisé habituellement aux États-Unis combinant carboplatine, etoposide et melphalan.
La toxicité aiguë principale de la combinaison Bu-Mel est la toxicité hépatique, maladie veino-occlusive du foie. Ce traitement induit en outre sur le long terme une stérilité pour les garçons et une défaillance ovarienne pour les filles qui conduit à proposer une cryopréservation ovarienne pour préserver les chances de fertilité et nécessite pour les filles la mise en place d’un traitement endocrinien substitutif pour déclencher la puberté. Le traitement local consiste en une chirurgie de la tumeur primitive dont l’objectif est d’obtenir une résection complète de la tumeur, ce qui est parfois complexe ou impossible en particulier pour les tumeurs médianes avec des rapports étroits avec les gros vaisseaux (aorte, tronc cœliaque, pédicules rénaux). Cette chirurgie est le plus souvent réalisée après la chimiothérapie d’induction et peut être décalée après la chimiothérapie à hautes doses en cas de difficultés opératoires identifiées. La radiothérapie est réalisée à la dose de 21 grays sur le volume tumoral avant la chirurgie. Le traitement d’entretien a pour objectif de contrôler la maladie résiduelle minime. Il comporte classiquement un traitement différenciant, l’acide rétinoïque, traitement oral donné en 2 prises quotidiennes à la dose totale de 160 mg/m2/j avec 6 cures de 14 jours espacées de 14 jours. Plus récemment le bénéfice de l’adjonction d’une immunothérapie a été démontré avec l’utilisation d’un anticorps monoclonal dirigé contre le ganglioside GD2 présent à la surface de la majorité des neuroblastomes. Il est classiquement administré 5 cures d’immunothérapie entre les cures d’acide rétinoïque.
Cet anticorps (dinutuximab bêta®) est utilisé seul ou avec d’autres cytokines dont le bénéfice est en cours d’étude. La survie à 5 ans des patients avec une maladie métastatique est maintenant de 50 % et doit continuer d’être améliorée. Les enfants avec une maladie localisée et une amplification de NMYC ont un pronostic moins sombre avec une survie sans événement de 75 % à 5 ans et un risque de rechute plus tardive nul. Les perspectives pour continuer d’améliorer ces résultats concernent différentes phases de traitement. La possibilité d’utiliser de façon plus précoce l’immunothérapie en combinaison avec de la chimiothérapie va être explorée prochainement du fait de résultats très encourageants observés chez des patients en rechute. Une autre piste est celle de l’utilisation d’une double chimiothérapie à hautes doses qui a été démontrée dans une étude du groupe américain COG comme permettant d’obtenir une survie supérieure à celle d’une seule chimiothérapie à hautes doses. Cette question est évaluée dans le protocole VERITAS pour les patients de très haut risque dans le groupe SIOPEN et le sera pour tous les patients de haut risque dans le prochain protocole.
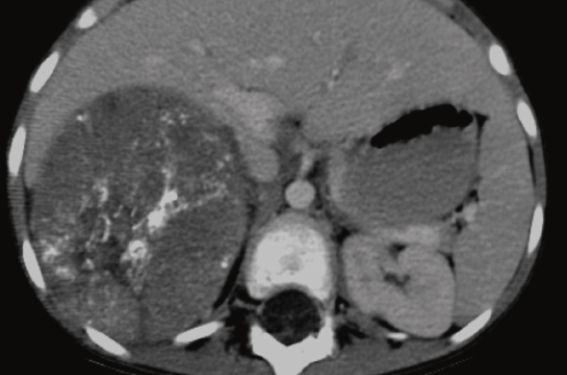
Figure 1. Neuroblastome surrénalien, masse hétérogène calcifiée. Pas de facteurs de risque chirurgicaux. Stade INRG L1.
Figure 2. Neuroblastome surrénalien droit, masse hétérogène dépassant la ligne médiane, décollant l’aorte du plan vertébral, englobant les vaisseaux médians. Stade INRG L2.
En conclusion
L’amélioration de la prise en charge thérapeutique des neuroblastomes passe par une amélioration de la compréhension de l’oncogenèse sur le plan génétique et épigénétique, du rôle du microenvironnement, et des relations entre l’hôte et la tumeur. L’apport d’une immunothérapie spécifique dans cette tumeur est une avancée importante dont les modalités d’administration et de combinaisons devront être explorée dans l’avenir pour améliorer le pronostic des formes de haut risque.
"Publié dans Pédiatrie Pratique"
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :

